Skandal bei Bandscheibenprothesen
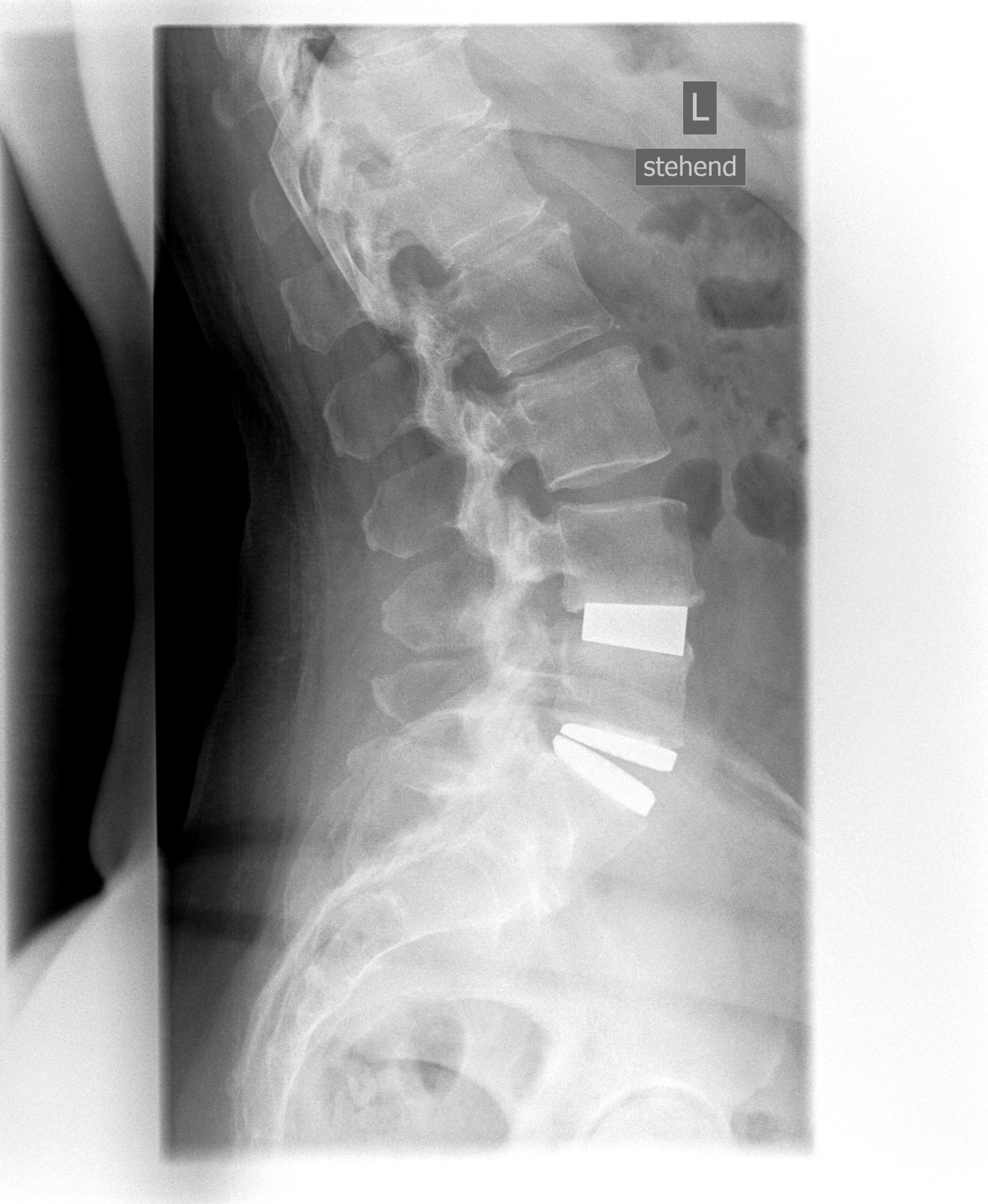 Ein neuer Skandal mit fehlerhaften Medizinprodukten betrifft tausende, wenn nicht hunderttausende Patienten weltweit. Die Firma Orthofix hat eine Bandscheibenprothese auf den Markt gebracht, die sich im Patienten auflösen und Wirbelsäule und Rückenmark schädigen kann. Betroffene können vom Hals abwärts gelähmt werden, andere leiden an starken Schmerzen und können ihren Alltag nicht mehr ohne fremde Hilfe bewältigen.
Ein neuer Skandal mit fehlerhaften Medizinprodukten betrifft tausende, wenn nicht hunderttausende Patienten weltweit. Die Firma Orthofix hat eine Bandscheibenprothese auf den Markt gebracht, die sich im Patienten auflösen und Wirbelsäule und Rückenmark schädigen kann. Betroffene können vom Hals abwärts gelähmt werden, andere leiden an starken Schmerzen und können ihren Alltag nicht mehr ohne fremde Hilfe bewältigen.
In Zusammenarbeit mit Betroffenen hat die Durom-SHG eine internationale Videoveranstaltung organisiert. Thema: „Medizinprodukteskandal bei Bandscheibenprothesen. Fallbeispiel: M6 von Spinal Kinetics/Orthofix“. Teilnehmende kamen unter anderem aus den USA, Kanada und Deutschland.