Körperverletzung durch fehlerhafte Hüftprothese?
Können Hersteller von Hüftprothesen oder Ärzte wegen gefährlicher Körperverletzung angeklagt werden, wenn sie eine fehlerhafte Hüftprothese auf den Markt bringen oder implantieren? Dieser Frage ging die Neue Westfälische Zeitung nach.
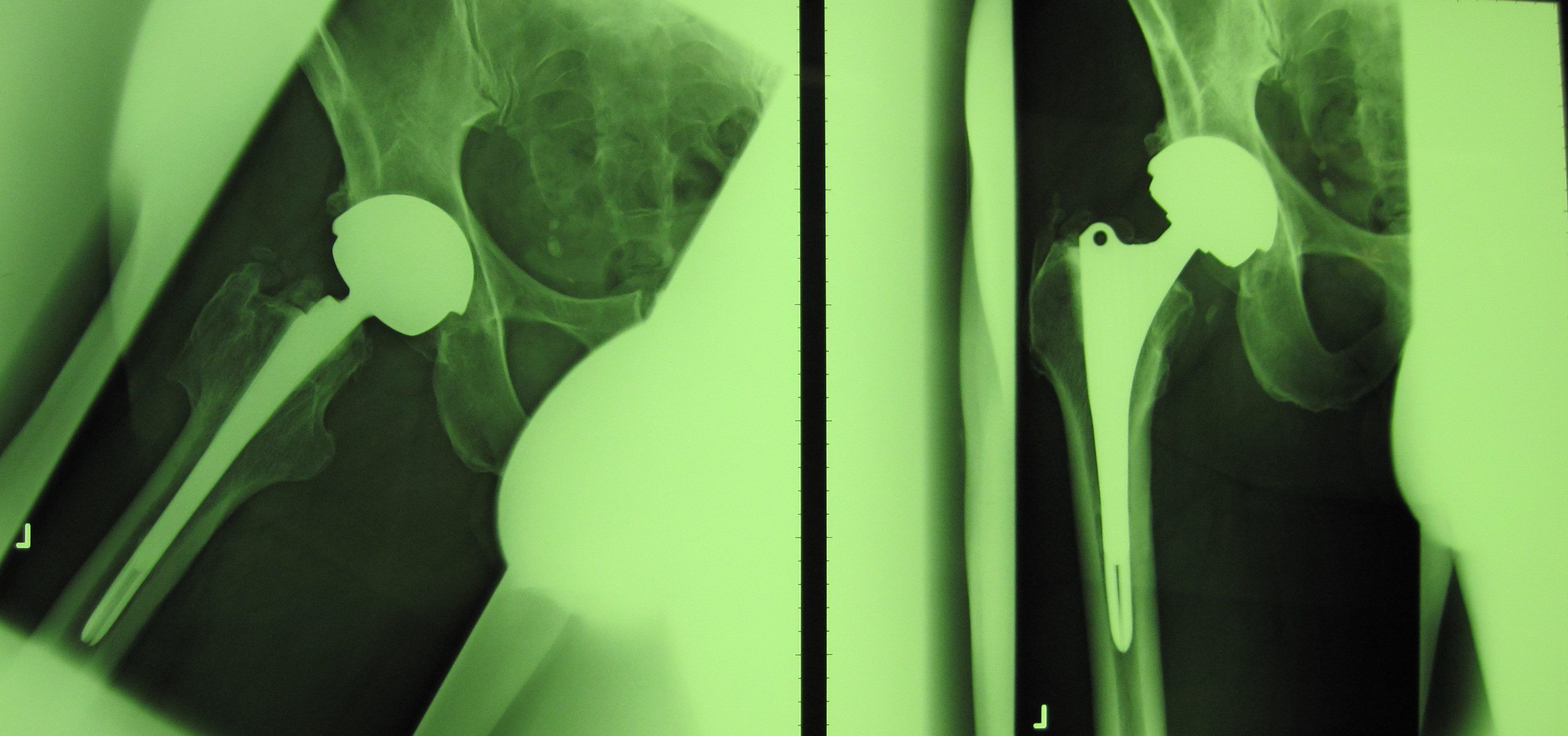
Die Neue Westfälische Zeitung berichtete am 06.03.2012 über Ermittlungen gegen einen leitenden Arzt des Püttlinger Knappschaftskrankenhaus. Ihm werde vorgeworfen, fehlerhafte Hüftprothesen der Firma DePuy auch dann noch implantiert zu haben, nachdem bereits bekannt war, dass diese fehlerhaft seien. Der Hersteller „DePuy Orthopaedics“ hatte für das Modell 2010 eine Rückrufaktion gestartet, nachdem bekannt geworden war, dass die Prothese bei zu vielen Patienten nach kurzer Zeit wieder ausgetauscht werden musste. Privatdozent Dr. Thomas Siebel soll an der Entwicklung der Prothesen mitgewirkt haben. Nach dem Bericht der NW ermittele die Staatsanwaltschaft Saarbrücken wegen des Verdachts der fahrlässigen Körperverletzung.
„Es gibt Hinweise, dass die Schäden länger bekannt waren, die Prothesen aber weiter vertrieben wurden“,
so der Sprecher der Staatsanwaltschaft Saarbrücken, Thomas Reinhardt.
Fehlerhafte Hüftprothese wird weiter implantiert
Chefarzt der Orthopädie und Unfallchirurgie im Knappschaftskrankenhauses (Stand 20.01.2026) ist nach wie vor Privatdozent Dr. Siebel.
„PD Dr. Thomas Siebel, Chefarzt der Orthopädie und Unfallchirurgie, ist ein überregional bekannter Operateur. Die von ihm mitentwickelten, modernsten Knie- und Hüftendoprothesen werden weltweit implantiert“
steht auf der Homepage des Krankenhauses.
Und weiter:
„Inzwischen haben wir unser Leistungsspektrum im Bereich Wirbelsäulenchirurgie weiter ausgebaut. Im Bereich Knie-Endoprothetik nutzen wir inzwischen die neueste robotikgestützte Technik mit dem Cori-System – einzigartig im Südwesten!
(Stand 20.01.2026).
Lückenhafte Kontrollen
Die ARD Sendung Panorama berichtete am 23.06.2015 um 21:15 Uhr unter dem Titel „Skandal um giftige Hüftprothesen“ über das fehlerhafte Prothesenmodell von DePuy. Auf der Webseite zur Sendung steht:
„Obwohl Hüftprothesen aus Metall krebserregende Gifte absonderten, hielten sich einige Produkte lange auf dem Markt. Vorwürfe richten sich gegen den Hersteller – und ein lückenhaftes Kontrollsystem“.
Hersteller wusste von Fehlerhaftigkeit
In der Sendung werden die Folgen der fehlerhaften Prothese beschrieben: Knochenschäden, Nekrosen und Tumore. Verursacht werden die gesundheitlichen Probleme durch erhöhten Abrieb von Chrom, Kobalt und Titan. Nicht nur gegen den Arzt des Püttlinger Kanppschaftskrankenhauses wurde von der Staatsanwaltschaft Saarbrücken ermittelt, sondern auch gegen den Geschäftsführer des Vertreibers im Saarland wegen „Inverkehrbringen gesundheitsgefährdender Medizinprodukte.“ In den Informationen des NDR zur Sendung steht:
„Die Strafverfolger werfen dem Vertreiber der künstlichen Metallhüften vor, von den Problemen gewusst und die Produkte dennoch weiter in Deutschland verkauft zu haben. Statistiken in Australien etwa hätten gezeigt, dass die Depuy-Prothesen seit 2005 eine „Revisionsrate über dem Durchschnitt“ aufwiesen.“
Nach Rechtsanwalt Jörg Heynemann aus Berlin, der viele der betroffenen DePuy Geschädigten vertritt, hätte das Produkt spätestens 2007 in Deutschland vom Markt genommen werden müssen. Aber erst 2010 startete DePuy einen „Rückruf“ in Deutschland. Dr. Siebel soll die fragliche Prothese noch länger in Patienten implantiert haben.
Warum wurde das Verfahren inzwischen eingestellt?
Wie die Volljuristin/Compliance-Beauftragte der Stabsstelle Recht und Compliance des Püttlinger Krankenhauses am 01.07.2024 mitteilt, wurde das Verfahren gegen den Chefarzt inzwischen eingestellt. Die Gründe der Einstellung werden nicht genannt.