Unabhängige Patientenberatung gefordert
Die Hüft-Skandale haben die Menschen verunsichert. Sie kommen mit ihren Fragen zur Durom-SHG. Es gibt zu wenig Unabhängige Patienteninformation.
Hier findet sich eine Aufstellung der Medienberichte über den durch die Verwendung der Durom-Metasul-LDH-Hüftprothesen verursachten Hüftprothesenskandal.
Die Hüft-Skandale haben die Menschen verunsichert. Sie kommen mit ihren Fragen zur Durom-SHG. Es gibt zu wenig Unabhängige Patienteninformation.
Seit vielen Jahren weisen wir als Selbsthilfegruppe immer wieder darauf hin, dass Metallabrieb, der durch künstliche Gelenke verursacht wird, gesundheitsschädlich ist. Die Sendung „Visite“ des NDR, die am Dienstag, 16. April 2019, um 20:15 Uhr ausgestrahlt wurde, bestätigt nun die von Metallabrieb im Körper ausgehende Gesundheitsgefahr für Patientinnen und Patienten mit einer Metall-auf-Metall Prothese.
Im Rahmen der aktuellen Berichterstattung hat das SWR Fernsehen in seinen aktuellen Sendungen am Montag, 08.06.2020 um 18:00 Uhr und um 19:30 Uhr ausführlich über das Urteil des Oberlandesgerichts Karlsruhe, Außenstelle Freiburg, berichtet.
Auf großes Interesse der Medien stieß die Verkündung der Entscheidung des OLG Karlsruhe in Freiburg in Sachen Durom-Metasul-LDH-Hüftprothese des Herstellers Zimmer Biomet, früher Zimmer. Das richtungsweisende Urteil gabe dem klagenden Patienten in allen Punkten Recht und bestätigte voll umfänglich das Urteil des Landgericht Freiburg (Aktenzeichen 1 O 240/10) vom 15.10.2018.
In zweiter Instanz wurde der Hersteller der fehlerhaften Durom-Hüftprothese dazu verurteilt,
Nach Auffassung der Gerichte hat Zimmer 2003 mit dem Durom-Metasul-LDH-Hüftprothesenmodell ein fehlerhaftes Prothesenmodell in Verkehr gebracht, obwohl
Das LG führt aus, dass ein Medizinprodukt nach § 3 Abs. 1 ProdHG dann einen Fehler aufweist, „wenn es nicht die Sicherheit bietet, die unter Berücksichtigung aller Umstände … berechtigterweise erwartet werden kann“. Dies sei hie der Fall. Außerdem habe zum Zeitpunkt der Markteinführung keine gültige Operationsanleitng vorgelegen, die auf die Probleme der Fügekraft des Konus hingewiesen hätte. Nach den nunmehr durchgeführten Tests und den vorliegenden Gutachten hätte hierzu eine Kraft aufgewendet werden müssen, die intraoperativ nicht erreichbar war. Der Operateur habe somit keine Chance gehabt, das Prothesenmodell so einzubauen, dass kein erhöhter und gesundheitsschädigender Metallabrieb entstehen kann.
Das Gericht stellt fest, dass der entstandene Metallabrieb erhöht und gesundheitlich bedenklich ist. Das Gericht führt aus, dass vieles darauf hindeute, dass Großkopfprothesen zu höheren Reibmomenten, Mikrobewegungen und damit einer höheren Belastung der Steckverbindung führen. Dies wusste der Hersteller bei Inverkehrbringung der Durom-Hüfte, unterließ notwendige und mögliche Tests jedoch.
Über die erste Verhandlung vor dem Oberlandesgericht gegen Zimmer am 13.01.2020 berichtete das Südwest Fernsehen. Doch die Prozesse ziehen sich in die Länge.
Am 20. November 2019 erschien in der Zeitschrift STERN NR. 37/2019 ein Artikel, der über den aktuellen Stand der Aktion aus der STERN Titelgeschichte vom 05.09.2019 berichtete. Veröffentlicht wurde ein Aufruf von über 130.000 Personen, die alle fordern: „So darf es nicht weiter gehen! Menschen vor Profit“ Unterzeichnet haben neben vielen einzelnen Ärzten, Präsidenten von Ärzteorganisationen und 19 Organisationen mit Verbindung zum Gesundheitswesen. Auf change.org/menschvorprofit wurde in Folge des STERN Berichts eine online Petition gestartet, welche bis zum 02.10.2019 bereits 52.000 Menschen unterstützen.
Der STERN griff damit eine von der SHG oft vorgebrachte Kritik an der Prothesenindustrie auf. Selbst Ärzte stellen immer wieder wirtschaftliche Interessen in den Vordergrund ihrer Behandlungsentscheidungen. Nach dem Motto „Viel hilft mir viel“ wird lieber eine Hüft-TEP zu viel als eine zu wenig auf den Weg gebracht. Bei der Dominanz von „Masse statt Klasse“ häuft sich zwangsläufig auch die Wahrscheinlichkeit, dass Fehler passieren. Vor allem bei der Kontrolle der Herstellerangaben zu einem neuen Medizinprodukt. Das Durom-Metasul-LDH-Hüftprothesenmodell der Firma Zimmer Biomet wurde als „der Mercedes der Hüftprothesen“ angepriesen. Manche haben den Versprechungen des Herstellers geglaubt, andere nicht.
Nach Auskunft der Geschäftsführung eines großen Herstellers von Medizinprodukten werden Medizinprodukte nach einer „firmeninternen Risikoanalyse“ auf den Markt gebracht. Die Frage drängt sich auf, welches Risiko die Firma prüft. Ist es das Risiko, für ein nicht ausreichend getestetes Prothesenmodell zur rechenschaft gezogen zu werden? Oder ist die Frage, wie sicher das Produkt für die Patiensten ist, die es implantiert bekommen sollen?
Dass das CE-Kennzeichen kein Qualitätssiegel ist und die Zulassungsverfahren in Europa keinerlei Sicherheit für die Patienten bietet, ist hinlänglich bekannt. Was also bedeutet „firmeninterne Risikoanalyse“? Wessen Risiko prüft die Firma? Das Risiko der Firma, bei einem möglicherweise fehlerhaften Medizinprodukt, welches sie auf den Markt zu bringen beabsichtigt, erwischt zu werden und am Ende zu Schadensersatz- und Schmerzensgeldzahlungen verurteilt zu werden? Oder wird das Risiko geprüft, mit dem Produkt die Entwicklungs- und Produktionskosten nicht erwirtschaften zu können? Dass das Risiko für Patienten geprüft wird, durch das Medizinprodukt einen gesundheitlichen Schaden zu erleiden, kann wohl als eher unwahrscheinlich abgetan werden.
Auszeichnung für die ImplantFiles. Die Journalisten der Implant Files wurden mit dem Award for Excellence in Health Care Journalism 2018 ausgezeichnet.
Ihre Rechercheergebnisse der ImplantFiles haben im Herbst 2018 die Verbraucher aufgeschreckt. Der internationale Rechercheverbund aus NDR, WDR und Süddeutscher Zeitung hatten erschreckende Beispiele von fehlerhaften Medizinprodukten, die in Patienten implantiert wurden, zusammen getragen. Als Hauptgrund für das Versagen der Implantate nennen die Journalisten das Konformitätsverfahren der EU. Bei diesem beauftragen die Hersteller eine sogenannte Benannte Stelle mit der Prüfung der Unterlagen. Die Unterlagen stammen ausschließlich vom Hersteller. Geprüft wird nicht, ob das Produkt Patienten gefährlich werden könnte, sondern nur, ob bei der Herstellung des Medizinproduktes die Vorgaben und Richtlinien der EU eingehalten wurden. Am Ende steht das CE-Kennzeichen, welches alle Produkte eingeholt werden muss, die in Europa verkauft werden sollen. Das CE-Kennzeichen gilt für Medizinprodukte genauso wie für Haushaltsgeräte, Spielwaren, etc..
Leider haben die Ergebnisse der ImplantFiles die verantwortlichen Entscheidungsträger nicht aufgeschreckt. Denn bis heute hat sich nicht viel bis gar nichts getan. Nichts-Tun hat Tradition: Schon vor zwölf Jahren sollten Operationen mit künstlichen Gelenken in einem bundesweiten Register erfasst werden. Doch bis heute gibt es außer der von den Herstellern als Prothesenregister verkauften Mogelpackung keine systematische und verpflichtende Erfassung der Implantate. Die Nichts-Tuer sitzen und saßen im Bundesgesundheitsministerium: vor 12 Jahren war Ulla Schmidt (SPD) Bundesgesundheitsministerin, dann kamen Philipp Rösler (FDP), Daniel Bahr (FDP) und Hermann Gröhe (CDU) und nun Jens Spahn. Alle waren offensichtlich nicht in der Lage, ein zentrales, unabhängiges und für mehr Patientensicherheit sorgendes Prothesenregister in Deutschland auf den Weg zu bringen.
Über die Gründe für diese Verweigerungshaltung kann man nur spekulieren. Liegt es daran, dass Medizinproduktehersteller mit erheblichem finanziellen Aufwand Lobbyarbeit betreiben? Liegt es daran, dass es ein Interessengeflecht zwischen Gesundheitsministerium und Medizinprodukteherstellern gibt? Liegt es daran, dass Patienten weder über Lobbyisten noch millionenschweren Etats verfügen? Liegt es daran, dass sich Patientensicherheit für Entscheidungsträger nicht „lohnt“?
Der aktuelle Bundesgesundheitsminister Jens Spahn hat nun wieder einmal die Einrichtung eines zentralen, beim DimDi (Deutsches Institut für Medizinische Dokumentation und Information) angesiedelten unabhängigen Prothesenregisters angekündigt: Herr Spahn, die Betroffenen warten darauf, dass Sie Worten nun endlich Taten folgen lassen!
Heftige Reaktionen gab es auf die Veröffentlichungen der ImplantFiles, welche große Sicherheitsmängel bei Medizinprodukten aufgedeckt hatten.
Die Dokumentation der Missstände, welche die Recherchegemeinschaft Süddeutsche Zeitung, NDR und WDR am 26.11.2018 veröffentlicht hatte, schlug hohe Wellen. Die Nachforschungen machten deutlich, dass das bisherige Zulassungssystem von Medizinprodukten in Deutschland und Europa nicht aufrecht erhalten werden darf. Denn es bietet keine Gewähr, dass die zugelassenen Produkte auch sicher sind für die Patienten. Wie Haushaltsgeräte und andere Produkte des Alltags, benötigen auch Medizinprodukte nur die CE-Kennzeichnung, damit sie in Europa verkauft werden dürfen. Die Beispiele, welche die ImplantFiles dokumentierten, zeigen zu welch ungeheuerlichen Exzessen dieses Verfahren führen kann. Ob Patienten dabei zu Schaden kommen oder gar ihr Leben verlieren, scheint niemanden zu interessieren.
Die wirtschaftliche Abhängigkeit der „Benannten Stellen“ von den Herstellern von Medizinprodukten verhindert eine effektive Kontrolle. Das ist so als ob ein Autobesitzer sein Fahrzeug in eine beliebige Werkstatt in Europa bringen kann, wenn er eine neue TÜV-Plakette braucht. Da die Werkstatt auch die nächste technische Untersuchung durchführen will, hat sie großes Interesse, den Auftraggeber nicht zu enttäuschen. Wenn sein Fahrzeug nicht wirklich verkehrssicher ist, kostet die Plakette etwas mehr.
Die Ergebnisse der Recherchegemeinschaft bestätigen die seit Jahren von der Selbsthilfegruppe Durom-Metasul-LDH-Hüftprothesen e.V. (SHG) erhobene Forderung nach unabhängigen und effektiveren Kontrollen für mehr Patientenschutz bei der Zulassung von Medizinprodukten.
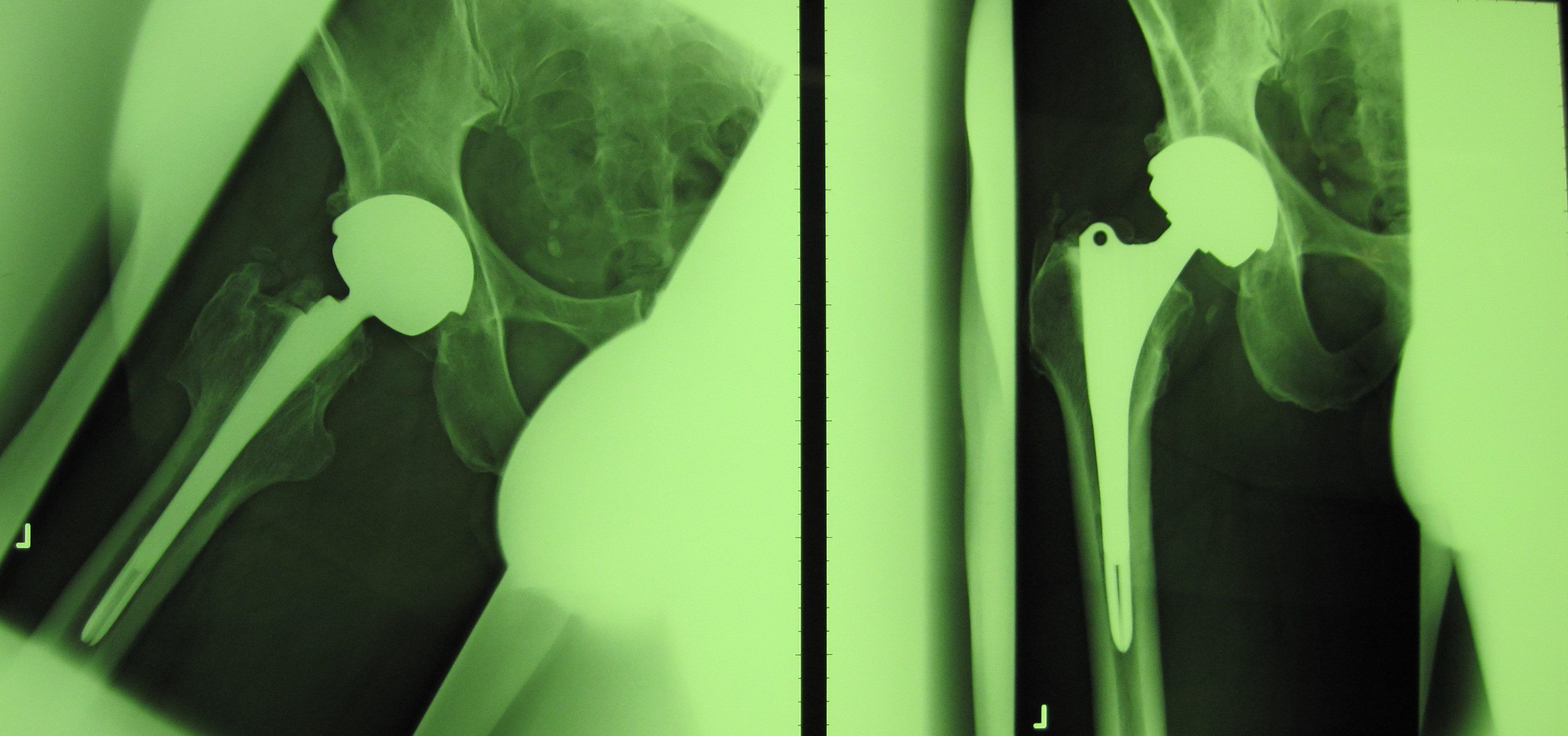
Die fehlerhafte Durom-Metasul-LDH-Hüftprothese der Firma Zimmer Biomet
Die SHG weist jedoch weiterhin darauf hin, dass auch die Überwachung der Medizinprodukte, nachdem sie auf den Markt gebracht wurden, verbessert werden muss. Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) ist zwar zuständig für Untersuchungen und Warnungen, wenn Probleme mit Medizinprodukten bekannt werden, ist aber abhängig davon, dass entsprechende Informationen und Unterlagen auch tatsächlich von Herstellern und/oder Kliniken eingereicht werden. Die „Empfehlung“, die das BfArM dann aussprechen kann, sollte die dann zuständige Landesbehörde zum Handel veranlassen. Geschieht auf Landesebene nichts, hat das BfArM keine Möglichkeit, aus seiner „Empfehlung“ eine „Anweisung“ zu machen. So blieb das bei den fehlerhaften Durom-Hüftprothesen der Firma Zimmer Biomet zuständige Regierungspräsidium Freiburg untätig und verweigerte die Umsetzung der Empfehlung des BfArM, das Produkt vom Markt zu nehmen. Begründet wurde das Nichts-tun damit, dass „eine juristische Auseinandersetzung mit dem Hersteller“ zu befürchten gewesen wäre“. Die Information der mit dem fehlerhaften Prothesenmodell belieferten Kliniken unterblieb, weil „sich die Probleme inzwischen ja rumgesprochen hätten“.
Neben der Verbesserung der Zulassungsverfahren für Medizinprodukte und ihrer Überwachung nach in Verkehrsbringung bedarf es eines unabhängigen und verpflichtenden Meldesystems der in den Menschen eingebrachten Medizinprodukte. Es kann nicht sein, dass auch das Bundesgesundheitsministerium keine Ahnung hat, welche Medizinprodukte wo mit welchem Erfolg in Deutschland auf dem Markt sind.
Metall-auf-Metall-Hüftprothesenmodelle mit einem Großkugelkopf werden von Experten inzwischen als problematisch angesehen. Die Gefahr, dass gesundheitsschädigender Metallabrieb entsteht, ist hoch. Die Erfahrungen der letzten Jahre zeigen, dass die erhofften Vorteile eines MoM Großkopfprothesenmodells nicht eingetreten seien. Der mechanisch-korrosiven Abrieb, schreibt Hannes A. Rüdiger in „Schweizerische Zeitschrift für Sportmedizin und Sporttraumatologie“ 61 (2), 39–42, 2013, hätte „lokale metall- assoziierte Weichteilreaktionen, Osteolysen und stark erhöhte systemische Ionen-Konzentrationen“ zur Folge gehabt, die zu teils erheblichen gesundheitlichen Einschränkungen bei Patienten geführt hätten. Weiterlesen
Jedes Medizinprodukt, welches in der EU zugelassen wird, muss von einer Benannten Stelle das CE-Kennzeichen vorweisen. Häufig wird der irreführende Eindruck erweckt, dass mit dem CE-Kennzeichen die Qualität des Produkts festgestellt wird. Dass dies jedoch weder bei Medizinprodukten der niedrigsten Risikoklasse wie Heftpflaster oder Scheren noch bei denen der höchsten Risikoklasse wie Herzschrittmachern oder Prothesen der Fall ist, beweisen die ZDF-Sendung „WISO“ und die Sendung „ODYSSO“ vom SWR.
Sie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Turnstile. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Facebook. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Instagram. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von X. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr Informationen