Das kann kein Zufall sein
Drohungen gegen die Durom-SHG nehmen seit dem vernichtenden Urteil gegen Zimmer Biomet zu. Will der Hersteller die Patientenvertretung einschüchtern?
Drohungen gegen die Durom-SHG nehmen seit dem vernichtenden Urteil gegen Zimmer Biomet zu. Will der Hersteller die Patientenvertretung einschüchtern?
In Deutschland und vielen anderen europäischen Ländern ist das Vertrauen in das Gesundheitssystem tief verankert. Doch wenn Medizinprodukte versagen oder Behandlungsfehler geschehen, beginnt für die betroffenen Patienten oft ein zermürbender Kampf – nicht nur um Gesundheit, sondern auch um Gerechtigkeit.
Zimmer Biomet war im August 2022 letztinstanzlich auch vor dem BGH mit seiner Behauptung, die Durom-Metasul-LDH-Hüftprothese sei nicht fehlerfrei, gescheitert. In der Folge wurde ein Teil der Patienten auf der Grundlage des rechtkräftigen OLG-Urteils vom 08.06.2020 mit EUR 25.000.- plus pauschal EUR 5.000.- Zinsen entschädigt. Alle anderen gingen bisher leer aus.
Bryan Hanson, Chef des amerikanischen Prothesenherstellers Zimmer Biomet, hat 2022 ein Jahresgehalt in Höhe von 16,4 Millionen US Dollar, umgerechnet 14.858.400.000,00 Euro eingestrichen. Gleichzeitig speist der Hersteller der fehlerhaften Durom-Hüftprothese betroffene Patienten mit 25.000.- EUR für ihr lebenslanges Leiden ab. Gipfel der Unverschämtheit: Eine ältere Patientin hatte rechts und links die fehlerhafte Durom-Hüftprothese implantiert bekommen. Sie musste wegen Metallosen, Osteolysen und nekrotischem Gewebe zweimal re-operiert werden. Eine weitere Operation wurde ihr von Chirurgen dringend angeraten. Doch durch die stzändigen Belastungen der Operationen ist ihr Herz geschädigt. Eine weitere Operation würde sie wohl nicht überleben. Ohne Rollator kann sie nun nicht einmal nehr kurze Strecken zurücklegen. Die Anwälte des Hersteller der Durom-Prothese haben ihr einen außergerichtlichen Vergleich vorgeschlagen: 5.000.- EUR. Nimmt sie diesen Vergleich nicht an, drohen die Anwälte mit einem jahrelangen Prozess.
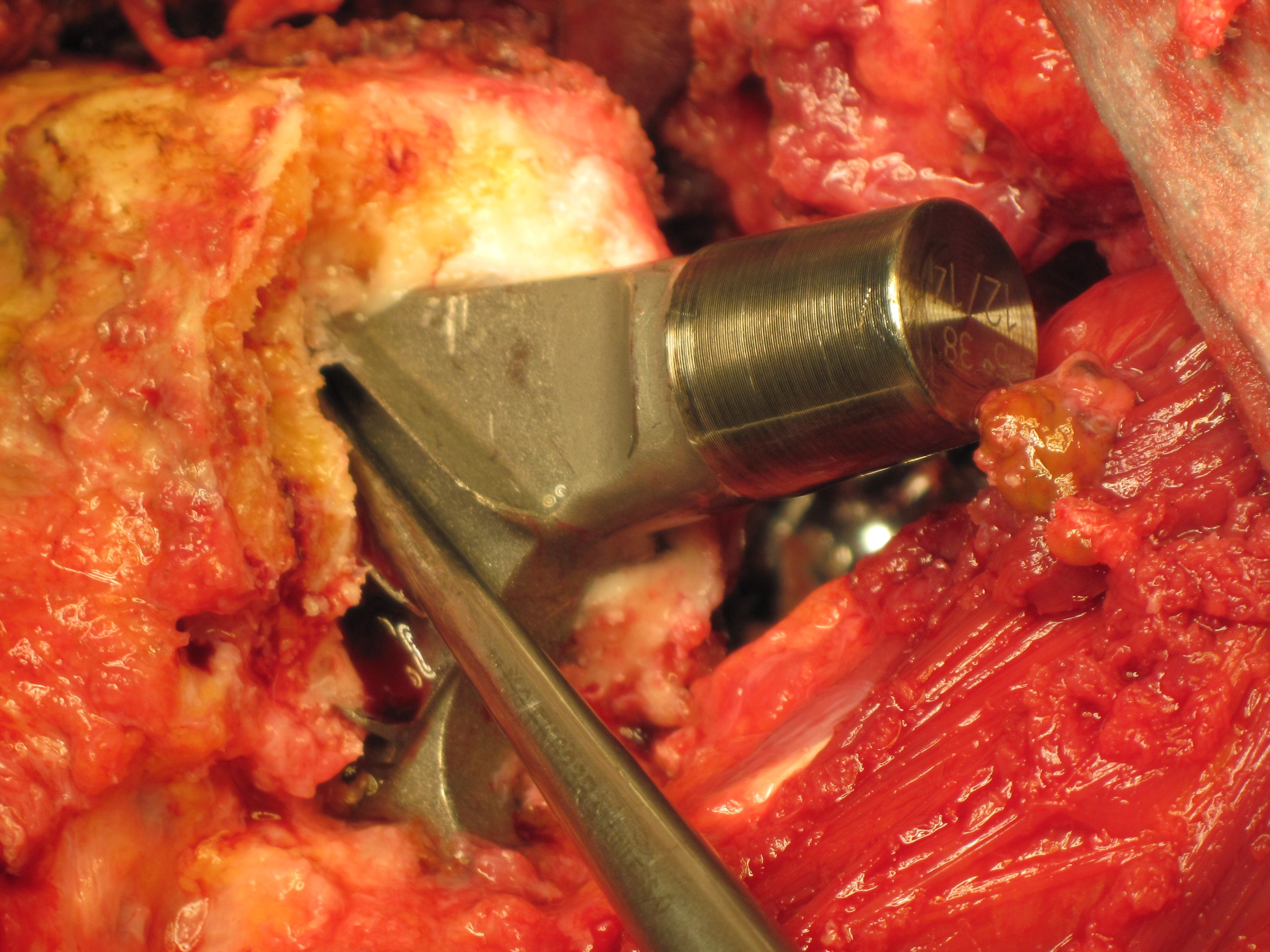
Durch erhöhten Metallabrieb verursachtes nekrotisches Gewebe
Am 9. August 2022 hat der Bundesgerichtshof nach über 12 Jahren das Verfahren eines Betroffenen gegen Zimmer Biomet wegen fehlerhafter Durom-Hüftprothese letztinstanzlich entschieden. Der Hersteller der Duom-Metasul-LDH-Hüftprothese wird zur Zahlung von Schadensersatz und Schmerzensgeld verurteilt, weil er die fehlerhafte Prothese ohne die nötigen Test durchzuführen, 2003 auf den Markt gebracht hatte. Allein in Freiburg litten über 1.000 Patienten nach der Implantation an den schmerzhaften Folgen des giftigen und krebserregenden Metallabriebs. Wegen Knochenkrebs, Metallosen, abgestorbenem und eitrigem Gewebe musste bei vielen deshalb in einer zweiten Operation die fehlerhafte Prothese entfernt werden. Bis heute leiden viele Betroffene unter den gesundheitlichen Folgen der Vergiftung.
Der 6. Senat des Bundesgerichtshofs hat am 9. August 2022 die Beschwerde von Zimmer Biomet zurückgewiesen. Das Oberlandesgericht (OLG) hatte dem Patienten Recht gegeben und die Firma zur Zahlung von Schmerzensgeld und Schadensersatz verurteilt. Revision gegen das Urteil hatte das OLG nicht zugelassen. Dagegen hatte sich die Beschwerde der Zimmer-Anwälte vor dem Bundesgerichtshof gerichtet.
Der Kläger hatte im Loretto Krankenhaus eine fehlerhafte Durom-Metasul-LDH-Hüftprothese erhalten. Die Anwälte der Firma hatten seit Prozessbeginn im Jahr 2010 versucht, mit allen juristischen Tricks ein rechtskräftiges Urteil zu verhindern. Erfolglos, wie sich nun feststellen lässt. Denn mit dem Beschluss des Bundesgerichtshofs dürfte das Urteil des OLG nun rechtskräftig werden.
Ob Rüstungsindustrie oder Medizinprodukteindustrie – in einem sind beide gleich: es geht um’s Geld. Ihre Produkte müssen an den Mann oder die Frau gebracht, Aktionäre zufrieden gestellt und die Gewinnspannen gesteigert werden. Dass der Mensch dabei auf der Strecke bleibt, interessiert nicht wirklich. Im Gegenteil: auf Kosten von Patienten werden getarnte Lobbyaktionen gestartet, um Medizinprodukte zu vermarkten. Davon berichtete Christian Fuchs am 6. Mai 2010 in DIE ZEIT, Nr. 19. Das ist zwar nun bereits viele Jahre her, aber die damaligen Recherchen von Christian Fuchs haben auch heute nichts an Aktualität verloren.
Stand 2021 sind alle bisherigen Gerichtsverfahren vor dem Landgericht Freiburg und dem Oberlandesgericht Karlsruhe zu Gunsten der klagenden Patienten entschieden worden. Die Anwälte der Firma Zimmer greifen zu allen juristischen Tricks, um die Verfahren in die Länge zu ziehen und rechtskräftige Urteile zu verhindern.
Da ist es nicht verwunderlich, dass die Entscheidung des OLG nun vor dem Bundesgerichtshof (BGH) gelandet ist. Das OLG hatte die Revision gegen sein Urteil nicht zugelassen, Dagegen hatten die Zimmer-Anwälte Beschwerde beim BGH) eingelegt. Das dürfte das Verfahren um weitere Jahre hinauszögern. Seriöse Anwälte geben der Beschwerde von Zimmer Biomet keine Chance. Offensichtlich verfolgt der Hersteller der fehlerhaften Duromprothese mit seiner Verzögerungstaktik andere Ziele: Zimmer Biomet setzt auf die „biologische Lösung“ und hofft, dass Patienten vor Ende ihres Verfahren versterben oder entkräftet aufgeben.
In einer Videokonferenz informierten Hanspeter Hauke und Manfred Mamber von der Durom-Selbsthilfegruppe (Durom-SHG) über den aktuellen Stand der gerichtlichen Verfahren gegen Zimmer Biomet.

Die fehlerhafte Durom-Metasul-LDH-Hüftprothese von Zimmer
Prof. Dr. Michael Morlock ist der Selbsthilfegruppe mehrfach durch seine seltsamen Behauptungen vor Gericht und seine Ausführungen in den Gutachten zur Durom-Metasul-LDH-Hüftprothese der Firma Zimmer aufgefallen. Die SHG hat sich deshalb mit einem Schreiben an die Deutsche Gesellschaft für Orthopädie und Orthopädische Chirurgie gewandt. Weiterlesen
 Bei der Bemessung von Schmerzensgeld vor Gerichten stellt sich regelmäßig die Frage, wie viel die Schmerzen des Patienten in EURO wert sind in Deutschland. Aus Amerika hört man immer wieder, dass Patienten nach einem erfolgreichen Gerichtsverfahren gegen Hersteller von Medizinprodukten Schmerzensgeld viele Millionen zugesprochen bekommen. Patienten-Millionäre sind in Deutschland bis heute keine bekannt geworden. Deutsche Gerichte orientieren sich an den seit Jahrzehnten nahezu unveränderten Summen, welche die immens gestiegenen Gewinne und die finanzielle Potenz der Hersteller unberücksichtigt lassen. Beträge von einigen tausend Euro für eine fehlerhafte Hüftprothese beispielsweise zahlen Weltmarktführer wie DePuy oder Zimmer aus der Portokasse. Einen Anreiz, neue Produkte umfassend und aufwändig vor Markteinführung zu testen, stellen sie nicht dar. Die Selbsthilfegruppe Durom-Metasul-LDH-Hüftprothesen fordert deshalb seit Jahren, Patienten in Verfahren gegen Hersteller solche Summen zuzusprechen, die eine abschreckende Wirkung haben. Weiterlesen
Bei der Bemessung von Schmerzensgeld vor Gerichten stellt sich regelmäßig die Frage, wie viel die Schmerzen des Patienten in EURO wert sind in Deutschland. Aus Amerika hört man immer wieder, dass Patienten nach einem erfolgreichen Gerichtsverfahren gegen Hersteller von Medizinprodukten Schmerzensgeld viele Millionen zugesprochen bekommen. Patienten-Millionäre sind in Deutschland bis heute keine bekannt geworden. Deutsche Gerichte orientieren sich an den seit Jahrzehnten nahezu unveränderten Summen, welche die immens gestiegenen Gewinne und die finanzielle Potenz der Hersteller unberücksichtigt lassen. Beträge von einigen tausend Euro für eine fehlerhafte Hüftprothese beispielsweise zahlen Weltmarktführer wie DePuy oder Zimmer aus der Portokasse. Einen Anreiz, neue Produkte umfassend und aufwändig vor Markteinführung zu testen, stellen sie nicht dar. Die Selbsthilfegruppe Durom-Metasul-LDH-Hüftprothesen fordert deshalb seit Jahren, Patienten in Verfahren gegen Hersteller solche Summen zuzusprechen, die eine abschreckende Wirkung haben. Weiterlesen
Sie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Turnstile. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Facebook. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Instagram. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von X. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr Informationen