Nach DUROM-Skandal: Reicht die MDR?
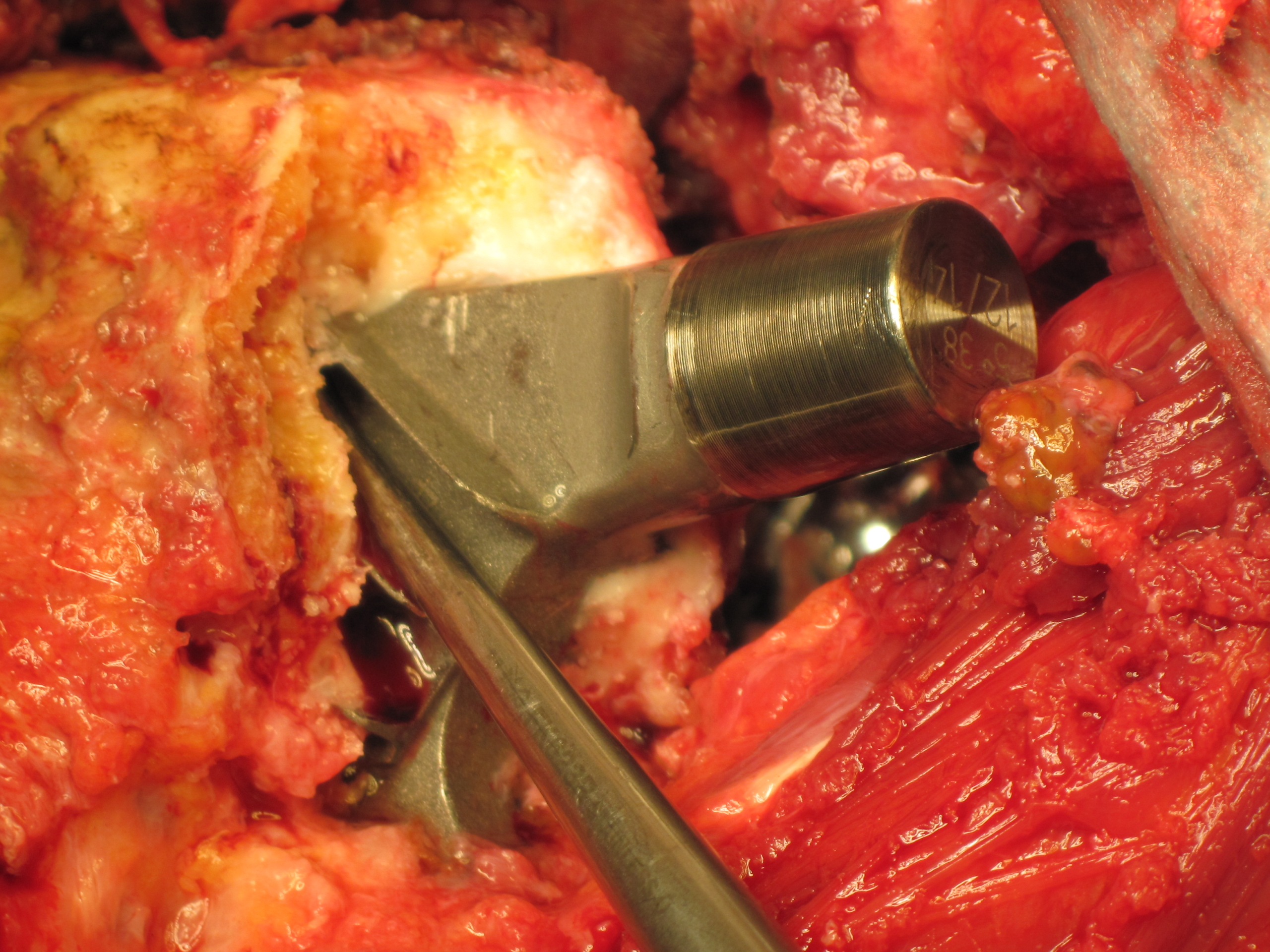
Die Durom-Metasul-LDH-Hüftprothese von Zimmer ist eine Fehlkonstruktion.
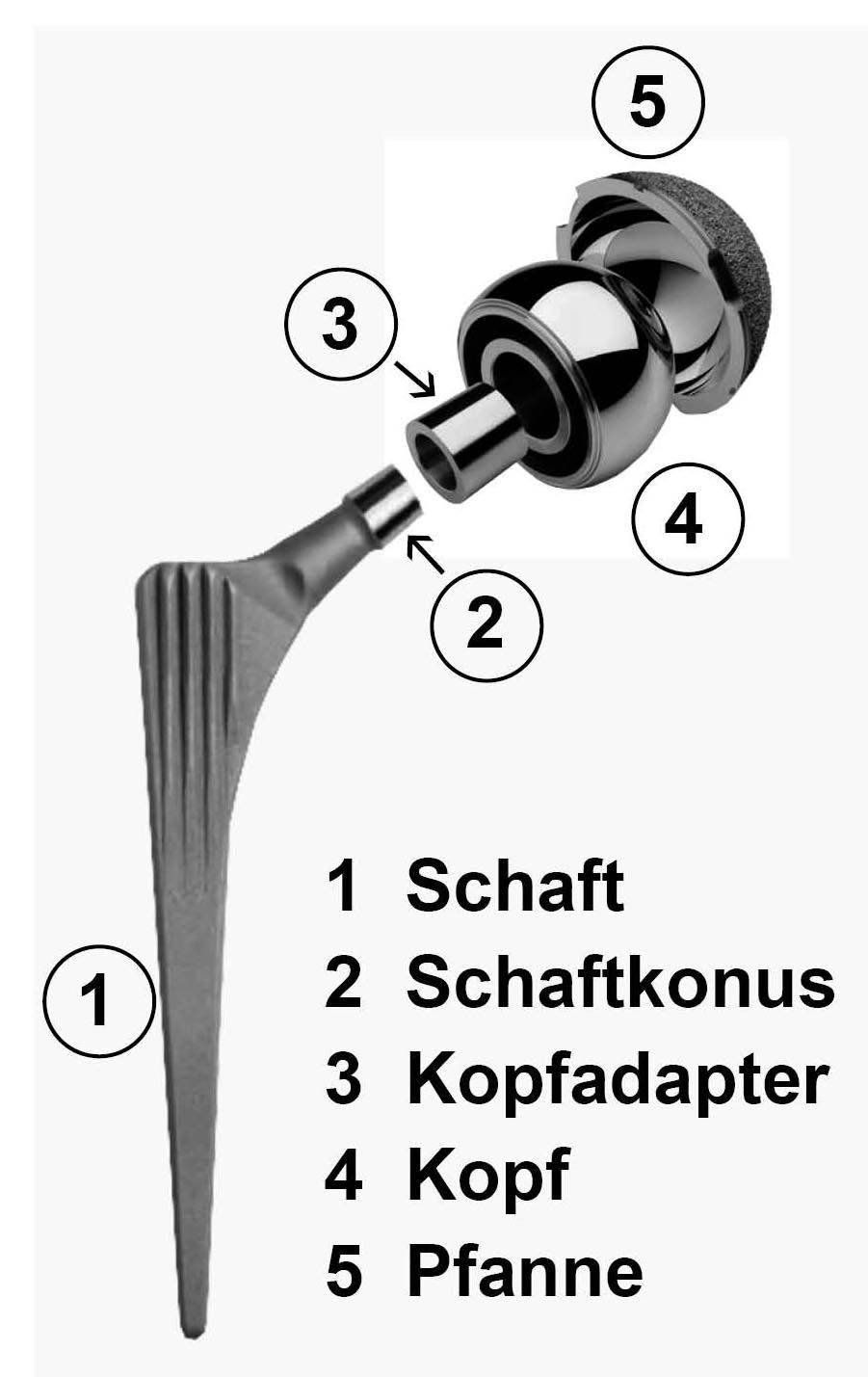
Mit der Medical Device Regulation (MDR) hat die Europäische Union die Vorschriften für Medizinprodukte deutlich verschärft. Anlass waren unter anderem Skandale um fehlerhafte Medizinprodukte, etwa die mangelhaften Brustimplantate des Herstellers PIP oder die Probleme mit Metall-auf-Metall-Hüftprothesen wie der DUROM-Prothese.
Ziel der MDR ist es, die Sicherheit der Patienten zu erhöhen und Medizinprodukte besser zu überwachen. Deshalb wurden viele Regelungen gegenüber der früheren Medical Devices Directive (MDD) deutlich verschärft. Dennoch sehen viele Fachleute weiterhin Schwachstellen.
Strengere klinische Nachweise
Früher konnten Hersteller häufig nachweisen, dass ihr Produkt einem bereits zugelassenen Medizinprodukt ähnlich war. Eigene klinische Studien waren deshalb oft nicht erforderlich.
Heute gelten strengere Regeln. Deshalb müssen Hersteller deutlich umfangreichere klinische Daten vorlegen. Gerade bei Hochrisikoimplantaten sind eigene klinische Untersuchungen heute wesentlich häufiger notwendig. Die bloße Berufung auf ein vergleichbares Produkt reicht in der Regel nicht mehr aus.