Medical Device Regulation
2012 begann die EU mit der Überarbeitung der EU-Regelungen und Richtlinien im Bereich Medizinprodukte. Nach vier Jahren wurden im Juni 2016 die Fassungen der neuen Verordnungen für Medizinprodukte (MDR) und In-Vitro-Diagnostika (IVDR) veröffentlicht. Waren die bisher gültigen Bestimmungen Regelungen sind MDR und IVDR nun Verordnungen, die ohne Ratifizierung der einzelnen Mitgliedsstaaten Rechtskraft erlangen. Mit der Inkraftsetzung durch die EU wird wird im ersten Halbjahr 2017 gerechnet.
Schweiz
Die Schweiz ist nicht Mitglied der EU. Die EU Verordnungen nicht, erlangen also auch nicht automatisch Gesetzeskraft in der Schweiz. Wollen die Schweizer Firmen jedoch weiterhin auf dem europäischen Markt aktiv sein, müssen die Verordnungen in nationales Recht überführt werden. Ansonsten drohen Handelshemmnisse und Einschränkungen beim Export aus der Schweiz in die EU. Mit einer Übernahme der EU-Verordnungen erhalten die Schweizer Firmen Zugang zu den Datenbanken der EU. Dies ist für sei ein nicht zu unterschätzender Vorteil.
Kritik angebracht
MDR und IVDR schränken die Nutzung des Äquivalenzprinzips ein und sehen mehr klinische Tests vor Markteinführung vor. Da jedoch nach wie vor an der Zulassungspraxis mit Benannten Stellen festgehalten wird, ist zu befürchten, dass auch künftig fehlerhafte Medizinprodukte auf den Markt kommen. Gefordert wird deshalb seit Jahren die Einrichtung einer unabhängigen Prüfstelle, die nicht vom Hersteller beauftragt und bezahlt wird. Auch darf sich deren Auftrag nicht darauf beschränken, die vom Hersteller eingereichten Unterlagen auf Vollständigkeit und Konformität mit den EU-Verordnungen zu prüfen. Geprüft werden muss vor allem, ob vom Medizinprodukt eine Gefährdung von Leib und Leben der Patienten ausgeht oder ausgehen kann und ob das Produkt eine innovative Neuerung darstellt, die medizinischen Fortschritt bei der Behandlung von Patienten bedeutet.
Herstellerinteressen vor Patientensicherheit
Bei Medizinprodukten der Risikoklassen Is, Im, IIa, IIb und III sowie In-Vitro Diagnostika gemäß Richtlinie 98/79/EG Anhang II muss für die Feststellung der Marktfähigkeit des Produkts eine Benannten Stelle bei der Konformitätsbewertung eingebunden werden. Durch MDR und IVDR werden zum Leidwesen der Hersteller einige Produkte in eine höhere Risikoklasse eingeordnet. Somit unterliegen sie neuerdings einem Zulassungsverfahren unter Einbeziehung einer Benannten Stelle. Dadurch wird das CE-Konformitätsverfahren für Hersteller für einige ihrer Produkte aufwändiger, doch ändert das verfahren nichts an der wirtschaftlichen Abhängigkeit der Benannten Stellen von den Medizinprodukteherstellern. Eine unabhängige Kontrollinstanz, die das Wirken der Benannten Stellen und der Hersteller kontrolliert, gibt es nach wie vor nicht.
Höhere Anforderungen an die Benannten Stellen
Auf der anderen Seite werden durch MDR und IVDR höhere Anforderungen an die Benannten Stellen gestellt. Das Johner Institut, eine Vereinigung zur Unterstützung der Hersteller bei den Marktzulassungsverfahren, nennt für die Benannten Stellen, abhängig vom Konformitätsbewertungsverfahren, das der Hersteller wählt, folgende Aufgaben der Benannten Stellen:
- Prüfung der technischen Dokumentation oder/und
- Prüfung jedes einzelnen Produkts oder/und
- Prüfung eines Baumusters oder/und
- Audits (auch unangekündigte), Zertifizierung und Überwachung des Qualitätsmanagementsystems.
Mit Inkrafttreten der Medical Device Regulation müssen sich die Benannten Stellen unter verschärften Voraussetzungen neu zertifizieren lassen. Dies wird dazu führen, dass viele auf der Strecke bleiben werden. Und die wenigen Neuen werden es schwer haben, den höheren Anforderungen mit entsprechend besser qualifiziertem Personal entsprechen zu können. Viele der bisher etwa 80 Benannten Stellen in Europa werden deshalb auf der Strecke bleiben, viele werden sich nicht erneuet bewerben. Das dürfte zu einem Engpass bei den Benannten Stellen führen. Im Juli 2018 gab es noch keine einzige Stelle, die benannt war. Die meisten hatten noch nicht einmal den Antrag gestellt!
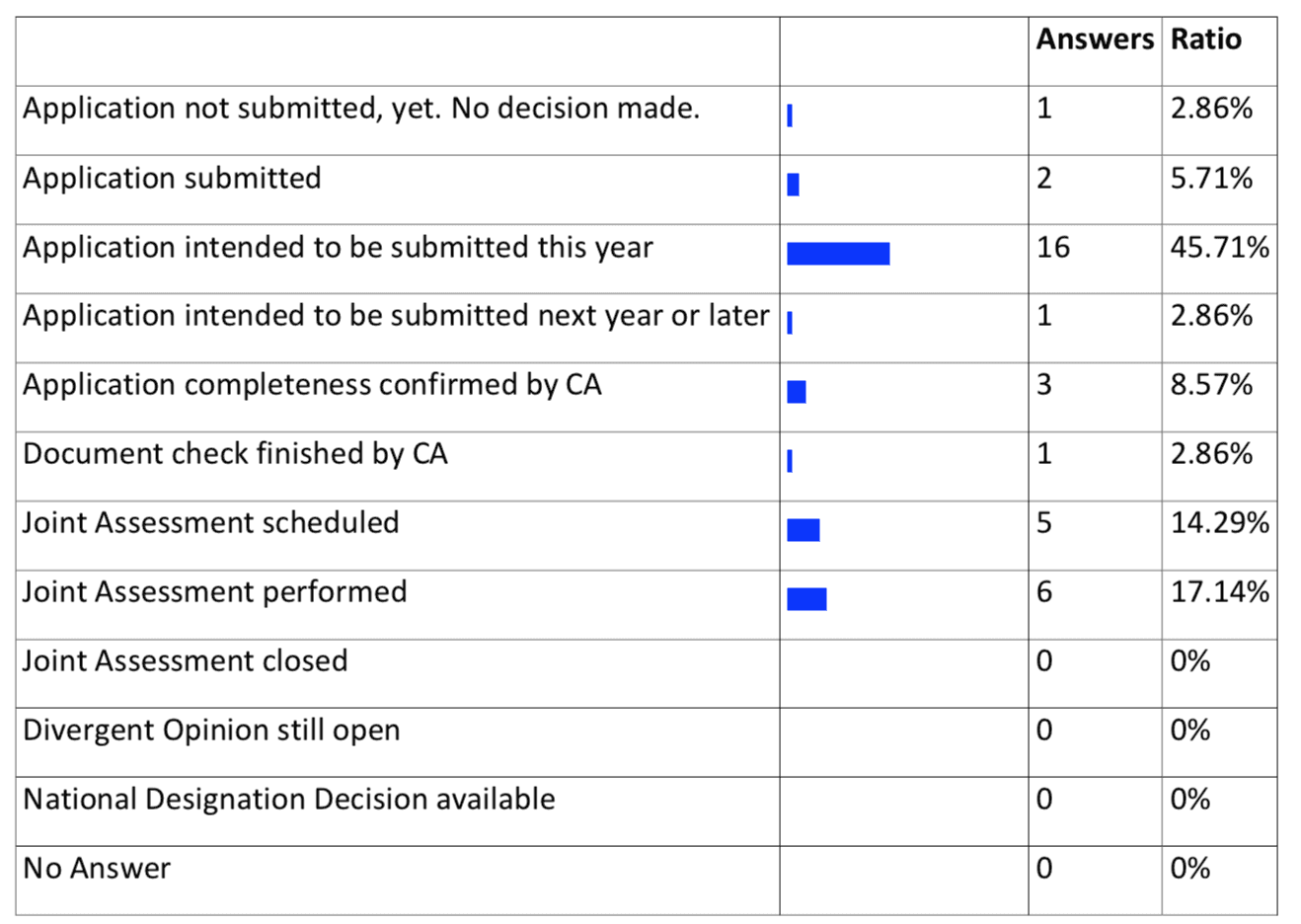
Benannte Stellen: Stand MDR Benennung MDR (Quelle)
Irrationale Überregulierung?
Hersteller werfen der EU vor, mit der MDR die Zulassungsverfahren in unverantwortlicher Weise zu über-regulieren. Eine Kritik, die immer dann vorgebracht wird, wenn von den Herstellern mehr Maßnahmen zur Erhöhung der Patientensicherheit abverlangt werden sollen. Aus Patientensicht ist das bei der MDR nicht der Fall. Dennoch führt die MDR nicht grundsätzlich zu mehr Patienten- und Produktsicherheit, solange das unsägliche System der Benannten Stellen nicht abgeschafft wird. An ihre Stelle muss eine unabhängige Prüfinstitution mit einem umfassenden Prüfauftrag treten (Quelle).
Hinterlasse einen Kommentar
An der Diskussion beteiligen?Hinterlasse uns deinen Kommentar!