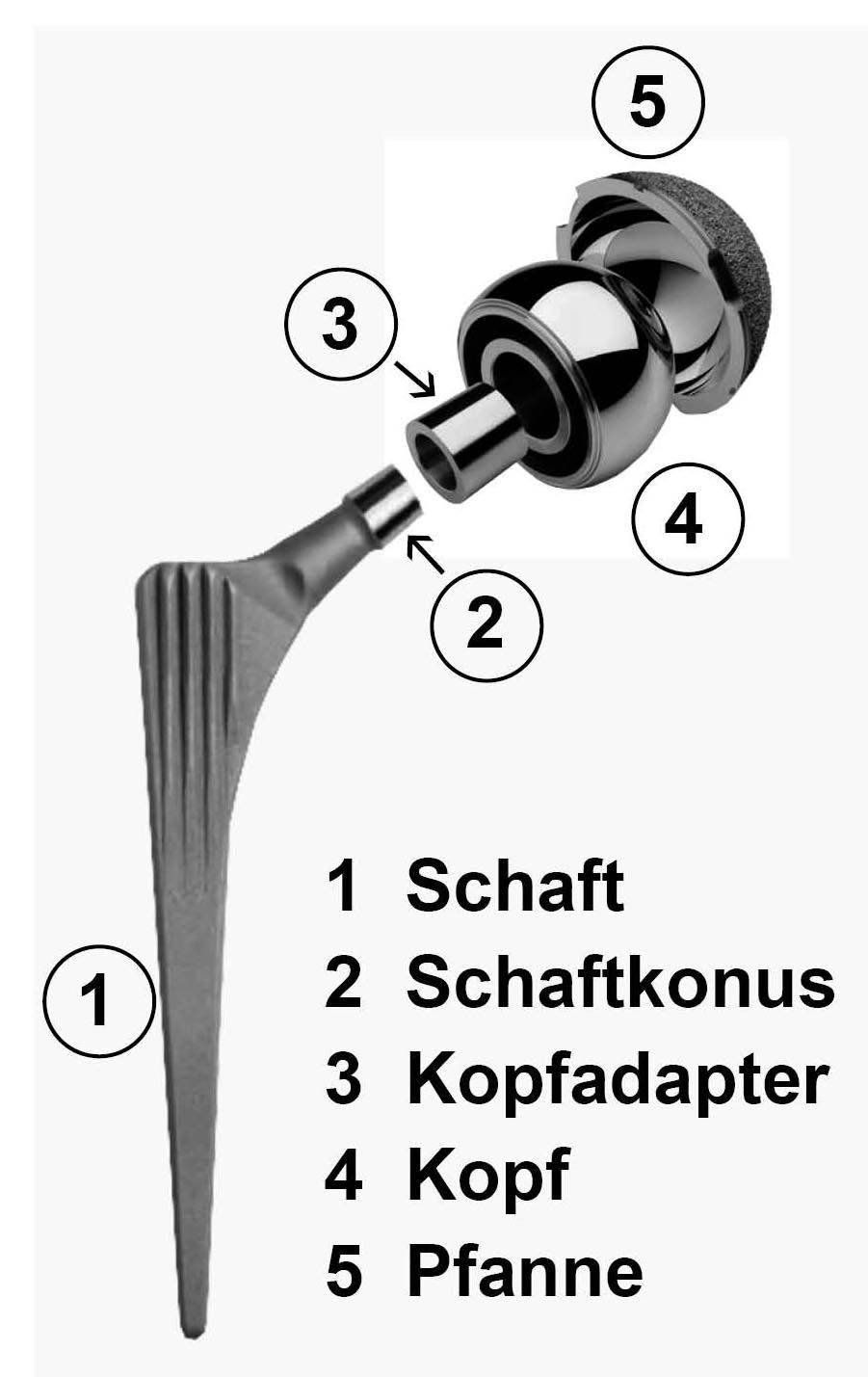
Sanfte OP-Methode bei Hüft-TEP: Die AMIS-Methode
Eine sanfte OP-Methode bei Hüft-TEP Operationen verspricht die sogenannte AMIS-Methode. Neben minimalinvasiven Operationstechniken, zu denen die AMIS-Methode zählt, gibt es die „klassische OP-Methode„, bei der der Operationsbereich großflächig frei gelegt wird und die computerunterstützte Operationsmethode, die zumindest teilweise von einem Computer gesteuert wird. Jede der genannten Methoden haben ihre Vor- und Nachteile, die vor jeder Hüft-TEP individuell und sorgfältig abgewogen werden muss.
AMIS-Methode
Nach den schlechten Erfahrungen mit ihrer Durom-Metasul-LDH-Hüftprothese von Zimmer Biomet ist eine Patientin bei ihrer zweiten Hüftprothese sehr viel kritischer vorgegangen. Nach ausführlicher Beratung und Recherchen auf der durom-hueftprobleme.de Homepage und im Internet, entschied sie sich mit ihrem Operateur für ein bewährtes Prothesenmodell. Auch eine andere Operationsmethode wurde gewählt: die AMIS-Methode (Anterior Minimally Invasive Surgery).
Erste Infos im Internet
Recherchiert man unter dem Stichwort „AMIS Methode“ stößt man überwiegend auf positive, ja teilweise fast euphorische Rückmeldungen von Patienten, die sich für diese Op-Methode entschieden hatten. Sie wird als die schonendste Methode für eine Hüftoperation beschrieben, weil keine Muskeln, Sehnen und Nerven auf dem Weg zum Hüftgelenk durchtrennt werden. Diese werden einfach auf die Seite geschoben. Der Zugang zum Operationsgebiet erfolgt minimal invasiv, also durch kleine Schnitte, mit denen der Operateur an den tiefer gelegenen Muskeln und Sehen vorbei zum Hüftgelenk gelangt. Dadurch müssen nach der Operation keine Muskel und Sehnen zusammenwachsen. Der Patient ist in wesentlich kürzerer Zeit wieder fit für den Alltag.
Vorteile der AMIS-Methode
Als weitere Vorteile der AMIS-Methode werden genannt:
- geringerer Blutverlust während der OP
- weniger Schmerzen und somit weniger Schmerzmittel nach der Hüft-TEP
- keine Schädigung der Hüft- und Oberschenkelmuskulatur
- Schonender Zugang zum Operationsgebiet
- schnellere Fitness nach der OP
- kürzerer Klinikaufenthalt
- kürzere Rehabilitationsphase
- kleinere Narbe
- reduziertes Luxationsrisiko
- geringere Gefahr des Hinkens
- weniger Langzeitschmerzen
Weitere Infos
Adressen für erste Informationen (keine Empfehlungen):
ECOM® Excellent Center of Medicine
Orthopädische Privatpraxis
Arabellastraße 17
81925 München
Tel: +49 89 92 333 94 – 0
Fax: +49 89 92 333 94 – 29
Mail: info@ecom-muenchen.de
Klinik für Orthopädie und Unfallchirurgie
Hugstetter Straße 55
79106 Freiburg
Prof. Dr. Lukas Konstantinidis
Sektionsleiter Endoprothetik
Telefon: 0761 270-61300
Email: lukas.konstantinidis@uniklinik-freiburg.de
Dr. med. Frank Burgbacher
Funktionsoberarzt
Telefon: 0761 270-61300
Email: frank.burgbacher@uniklinik-freiburg.de