Heftige Reaktionen gab es auf die Veröffentlichungen der ImplantFiles, welche große Sicherheitsmängel bei Medizinprodukten aufgedeckt hatten.
Die Dokumentation der Missstände, welche die Recherchegemeinschaft Süddeutsche Zeitung, NDR und WDR am 26.11.2018 veröffentlicht hatte, schlug hohe Wellen. Die Nachforschungen machten deutlich, dass das bisherige Zulassungssystem von Medizinprodukten in Deutschland und Europa nicht aufrecht erhalten werden darf. Denn es bietet keine Gewähr, dass die zugelassenen Produkte auch sicher sind für die Patienten. Wie Haushaltsgeräte und andere Produkte des Alltags, benötigen auch Medizinprodukte nur die CE-Kennzeichnung, damit sie in Europa verkauft werden dürfen. Die Beispiele, welche die ImplantFiles dokumentierten, zeigen zu welch ungeheuerlichen Exzessen dieses Verfahren führen kann. Ob Patienten dabei zu Schaden kommen oder gar ihr Leben verlieren, scheint niemanden zu interessieren.
Die wirtschaftliche Abhängigkeit der „Benannten Stellen“ von den Herstellern von Medizinprodukten verhindert eine effektive Kontrolle. Das ist so als ob ein Autobesitzer sein Fahrzeug in eine beliebige Werkstatt in Europa bringen kann, wenn er eine neue TÜV-Plakette braucht. Da die Werkstatt auch die nächste technische Untersuchung durchführen will, hat sie großes Interesse, den Auftraggeber nicht zu enttäuschen. Wenn sein Fahrzeug nicht wirklich verkehrssicher ist, kostet die Plakette etwas mehr.
Die Ergebnisse der Recherchegemeinschaft bestätigen die seit Jahren von der Selbsthilfegruppe Durom-Metasul-LDH-Hüftprothesen e.V. (SHG) erhobene Forderung nach unabhängigen und effektiveren Kontrollen für mehr Patientenschutz bei der Zulassung von Medizinprodukten.
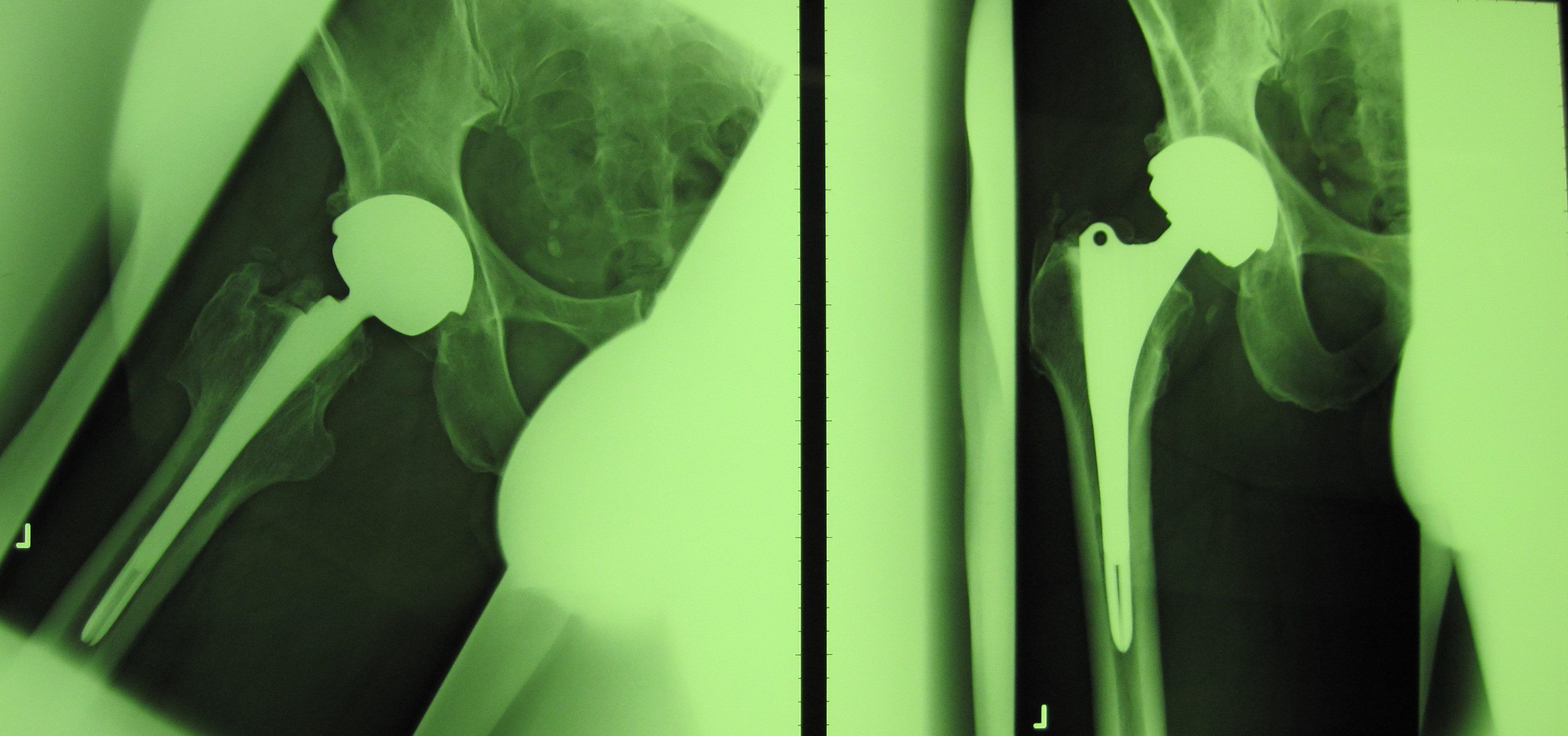
Die fehlerhafte Durom-Metasul-LDH-Hüftprothese der Firma Zimmer Biomet
Die SHG weist jedoch weiterhin darauf hin, dass auch die Überwachung der Medizinprodukte, nachdem sie auf den Markt gebracht wurden, verbessert werden muss. Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) ist zwar zuständig für Untersuchungen und Warnungen, wenn Probleme mit Medizinprodukten bekannt werden, ist aber abhängig davon, dass entsprechende Informationen und Unterlagen auch tatsächlich von Herstellern und/oder Kliniken eingereicht werden. Die „Empfehlung“, die das BfArM dann aussprechen kann, sollte die dann zuständige Landesbehörde zum Handel veranlassen. Geschieht auf Landesebene nichts, hat das BfArM keine Möglichkeit, aus seiner „Empfehlung“ eine „Anweisung“ zu machen. So blieb das bei den fehlerhaften Durom-Hüftprothesen der Firma Zimmer Biomet zuständige Regierungspräsidium Freiburg untätig und verweigerte die Umsetzung der Empfehlung des BfArM, das Produkt vom Markt zu nehmen. Begründet wurde das Nichts-tun damit, dass „eine juristische Auseinandersetzung mit dem Hersteller“ zu befürchten gewesen wäre“. Die Information der mit dem fehlerhaften Prothesenmodell belieferten Kliniken unterblieb, weil „sich die Probleme inzwischen ja rumgesprochen hätten“.
Neben der Verbesserung der Zulassungsverfahren für Medizinprodukte und ihrer Überwachung nach in Verkehrsbringung bedarf es eines unabhängigen und verpflichtenden Meldesystems der in den Menschen eingebrachten Medizinprodukte. Es kann nicht sein, dass auch das Bundesgesundheitsministerium keine Ahnung hat, welche Medizinprodukte wo mit welchem Erfolg in Deutschland auf dem Markt sind.